当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Revisiting Textbook Azide-Clock Reactions: A “Propeller-Crawling” Mechanism Explains Differences in Rates
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2024-04-30 , DOI: 10.1021/jacs.4c03360 Anthony T. Bogetti 1 , Matthew C. Zwier 2 , Lillian T. Chong 1
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2024-04-30 , DOI: 10.1021/jacs.4c03360 Anthony T. Bogetti 1 , Matthew C. Zwier 2 , Lillian T. Chong 1
Affiliation
|
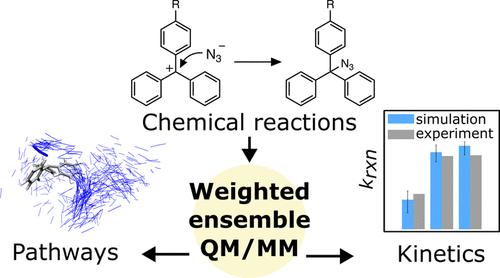
An ongoing challenge to chemists is the analysis of pathways and kinetics for chemical reactions in solution, including transient structures between the reactants and products that are difficult to resolve using laboratory experiments. Here, we enabled direct molecular dynamics simulations of a textbook series of chemical reactions on the hundreds of ns to μs time scale using the weighted ensemble (WE) path sampling strategy with hybrid quantum mechanical/molecular mechanical (QM/MM) models. We focused on azide-clock reactions involving addition of an azide anion to each of three long-lived trityl cations in an acetonitrile–water solvent mixture. Results reveal a two-step mechanism: (1) diffusional collision of reactants to form an ion-pair intermediate; (2) “activation” or rearrangement of the intermediate to the product. Our simulations yield not only reaction rates that are within error of experiment but also rates for individual steps, indicating the activation step as rate-limiting for all three cations. Further, the trend in reaction rates is due to dynamical effects, i.e., differing extents of the azide anion “crawling” along the cation’s phenyl-ring “propellers” during the activation step. Our study demonstrates the power of analyzing pathways and kinetics to gain insights on reaction mechanisms, underscoring the value of including WE and other related path sampling strategies in the modern toolbox for chemists.
中文翻译:

重温教科书叠氮化物时钟反应:“螺旋桨爬行”机制解释了速率差异
化学家面临的一个持续挑战是分析溶液中化学反应的路径和动力学,包括反应物和产物之间难以通过实验室实验解决的瞬态结构。在这里,我们使用加权系综 (WE) 路径采样策略和混合量子力学/分子力学 (QM/MM) 模型,在数百纳秒到微秒的时间尺度上对教科书上的一系列化学反应进行了直接分子动力学模拟。我们重点研究叠氮时钟反应,涉及在乙腈-水溶剂混合物中向三个长寿命三苯甲基阳离子中的每一个添加叠氮阴离子。结果揭示了两步机制:(1)反应物的扩散碰撞形成离子对中间体; (2)中间体“活化”或重排为产物。我们的模拟不仅产生了实验误差范围内的反应速率,而且还产生了各个步骤的速率,表明活化步骤是所有三种阳离子的速率限制。此外,反应速率的趋势是由于动力学效应,即在活化步骤期间叠氮化物阴离子沿着阳离子的苯环“螺旋桨”“爬行”的不同程度。我们的研究证明了分析路径和动力学以深入了解反应机制的力量,强调了将 WE 和其他相关路径采样策略纳入化学家现代工具箱中的价值。
更新日期:2024-04-30
中文翻译:
重温教科书叠氮化物时钟反应:“螺旋桨爬行”机制解释了速率差异
化学家面临的一个持续挑战是分析溶液中化学反应的路径和动力学,包括反应物和产物之间难以通过实验室实验解决的瞬态结构。在这里,我们使用加权系综 (WE) 路径采样策略和混合量子力学/分子力学 (QM/MM) 模型,在数百纳秒到微秒的时间尺度上对教科书上的一系列化学反应进行了直接分子动力学模拟。我们重点研究叠氮时钟反应,涉及在乙腈-水溶剂混合物中向三个长寿命三苯甲基阳离子中的每一个添加叠氮阴离子。结果揭示了两步机制:(1)反应物的扩散碰撞形成离子对中间体; (2)中间体“活化”或重排为产物。我们的模拟不仅产生了实验误差范围内的反应速率,而且还产生了各个步骤的速率,表明活化步骤是所有三种阳离子的速率限制。此外,反应速率的趋势是由于动力学效应,即在活化步骤期间叠氮化物阴离子沿着阳离子的苯环“螺旋桨”“爬行”的不同程度。我们的研究证明了分析路径和动力学以深入了解反应机制的力量,强调了将 WE 和其他相关路径采样策略纳入化学家现代工具箱中的价值。




























 京公网安备 11010802027423号
京公网安备 11010802027423号